De Mucopolysaccharidose is een verzamelnaam voor lysosomale stapelingsziekten die zijn gebaseerd op de opslag van glycosaminoglycanen. Alle ziekten ontwikkelen vergelijkbare symptomen en vormen. De ernst van de syndromen loopt sterk uiteen.
Wat is mucopolysaccharidose?

© Sebastian Kaulitzki - stock.adobe.com
EEN Mucopolysaccharidose er bestaat niet zoiets als een enkele ziekte. De term mucopolysaccharidose is een verzamelnaam voor een groot aantal stapelingsziekten, die zijn gebaseerd op opslagstoornissen van glycosaminoglycanen (GAG) in de lysosomen van cellen. De opslag vindt progressief plaats omdat het verbreken van de verbindingen niet lukt.
$config[ads_text1] not found
Alle mucopolysaccharidoses zijn genetisch bepaald. Bij elke ziekte ontbreekt een specifiek enzym dat de afbraak van de overeenkomstige GAG katalyseert. Alle mucopolysaccharidoses zijn zeer zeldzame ziekten en vertonen vaak een vergelijkbaar verloop. Indien onbehandeld, vernietigen de steeds toenemende afzettingen de cellen. De orgels worden daarbij vernietigd. De ziekte kan zowel in de kindertijd als op volwassen leeftijd beginnen.
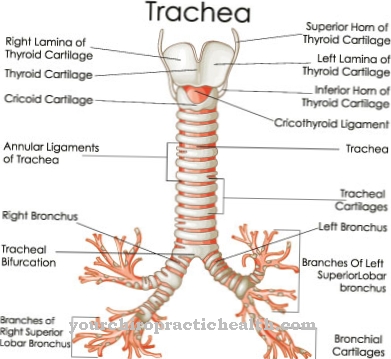
Mucopolysaccharidose kan worden veroorzaakt door vier verschillende groepen glycosaminoglycanen:
- Heparansulfaat
- Keratansulfaat
- Chondroïtinesulfaat
- Dermatansulfaat.
Alle glycosaminoglycanen bestaan uit een polysaccharideketen die aan een eiwit is bevestigd. De koolhydraatcomponent maakt 95 procent uit en de eiwitcomponent vijf procent van de molecuulmassa. Afhankelijk van welke glycosaminoglycaan en welk enzym wordt aangetast, kunnen mucopolysaccharidosen worden onderverdeeld in zes verschillende hoofdvormen: deze omvatten de ziekte van Hurler / Scheie (MPS I), de ziekte van Hunter (MPS II), de ziekte van Sanfilippo (MPS III), de ziekte van Morquio ( MPS IV), de ziekte van Maroteaux-Lamy (MPS VI) en de ziekte van Sly (MPS VII). Alle soorten hebben ernstige en milde vormen.
$config[ads_text2] not foundoorzaken
De oorzaak van alle mucopolysaccharidosen is een toenemende opslag van glycosaminoglycanen (GAG's) in de lysosomen van de cellen. De afbraak van de overeenkomstige biopolymeren wordt verstoord. Voor elke individuele aandoening ontbreekt óf een bepaald enzym of dit enzym werkt niet goed. Er kunnen verschillende mutaties zijn voor elk enzym. De overerving van de overeenkomstige mutatie kan autosomaal recessief, autosomaal dominant of x-gebonden recessief zijn.
Omdat een enzymatisch proces meestal meerdere reactiestappen omvat, kunnen theoretisch verschillende enzymen worden gemuteerd voor hetzelfde glycosaminoglycaan. De symptomen van de aandoening zouden hetzelfde of vergelijkbaar zijn.
- Bij MPS I, De ziekte van Hurler of Scheie, is het enzym alfa-l-iduronidase defect.
- MPS II vertegenwoordigt het Hunters-syndroom met een defecte iduronaat-2-sulfatase.
- Het Sanfilippo-syndroom (MPS III) kunnen worden onderverdeeld in verschillende subtypen. Bij deze aandoening kunnen verschillende enzymen worden aangetast.
- Ziekte van Morquio (MPS IV) wordt veroorzaakt door een defecte β-galactosidase.
- Bij het Maroteaux-Lamy-syndroom (MPS VI) het is N-acetyl-galactosamine-4-sulfaat sulfatase.
- Ziekte van Sly (MPS VII) wordt veroorzaakt door defecte β-glucuronidase. Wanneer de overeenkomstige glycosaminoglycanen worden opgeslagen in de lysosomen, worden deze groter en groter.
De cellen worden ook groter omdat ze steeds meer ruimte nodig hebben voor niet-afgebroken GAG's. Dit is ook merkbaar bij de vergroting van veel organen. Een typisch symptoom is de constante vergroting van de lever en milt. Indien onbehandeld, leiden de stapelingsziekten tot de dood door de geleidelijke vernietiging van de organen.
$config[ads_text3] not foundSymptomen, aandoeningen en tekenen
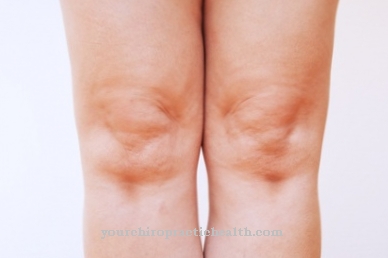
De symptomen zijn bij alle ziekten gelijk. Er zijn ernstige en milde vormen. Een mild beloop betekent echter alleen dat de ziekte langzamer verloopt. De laatste cursus is altijd hetzelfde. Er is progressieve vervorming van het skelet, gewrichtscontracturen, verruwing van gelaatstrekken en vergroting van de lever en milt.
De mentale en motorische vaardigheden nemen af op korte of lange termijn. Bij de ernstige vormen van de aandoeningen lijken de klinische beelden sterk op elkaar. Navelstreng- en liesbreuken, hartproblemen en luchtweginfecties komen in een vroeg stadium voor. Na verloop van tijd ontwikkelen de vernauwing van de luchtwegen en de vergroting van de amandelen en amandelen zich tot enorme slaapapneu-problemen.
Diagnose en verloop van de ziekte
Mucopolysaccharidosen kunnen worden gediagnosticeerd door de urine te onderzoeken op uitgescheiden glycosaminoglycanen. Bij mucopolysaccharidose worden de waarden altijd verhoogd. De activiteit van het vermoedelijke defecte enzym in de leukocyten of fibroblasten kan ook worden bepaald. Een bepaald patroon van uitscheiding van de glycosaminoglycanen leidt het vermoeden tot een overeenkomstig enzym, dat vervolgens wordt onderzocht.
Complicaties
Door mucopolysaccharidose lijden de getroffenen aan verschillende misvormingen en skeletklachten. Er treden vervormingen op die het dagelijks leven van de patiënt aanzienlijk kunnen beperken. In de regel worden de gewrichten ook aangetast door mucopolysaccharidose, zodat de beweging van de patiënt wordt beperkt.
Vooral kinderen worden getroffen en hebben een ernstig vertraagde ontwikkeling, zodat ook op volwassen leeftijd diverse gevolgschade kan optreden. Het is niet ongebruikelijk dat mucopolysaccharidose problemen met het hart of de ademhaling veroorzaakt. In het ergste geval kan plotselinge hartdood leiden tot het overlijden van de betrokken persoon. Door de ademhalingsmoeilijkheden lijden patiënten aan vermoeidheid en vermoeidheid.
$config[ads_text4] not foundOok de veerkracht van getroffenen neemt enorm af. Het is niet ongebruikelijk dat ademhalingsmoeilijkheden 's nachts leiden tot slaapproblemen en dus tot depressie. De kwaliteit van leven van de patiënt wordt aanzienlijk verminderd door de mucopolysaccharidose. Een causale behandeling van deze ziekte is helaas niet mogelijk. De getroffenen zijn daarom afhankelijk van beenmergdonoren om de symptomen te behandelen. Er zijn geen bijzondere complicaties. In de meeste gevallen zijn patiënten echter afhankelijk van levenslange therapie.
Wanneer moet je naar de dokter gaan?
Veranderingen en afwijkingen in de lichaamsstructuur duiden op een aantasting van de gezondheid. Een doktersbezoek is noodzakelijk zodra er blijvende optische bijzonderheden zijn of de betrokkene moeite heeft met het opzettelijk optimaliseren van zijn houding.Zwelling van de gewrichten, veranderingen in gelaatstrekken of vergroting van de borst moeten intensief door een arts worden onderzocht, zodat een diagnose kan worden gesteld. Een arts is vereist als er beperkingen zijn in de bewegingsmogelijkheden, onregelmatigheden in de dagelijkse vrijwillige controle en een afname van fysieke en mentale prestaties. Raadpleeg een arts bij verstoringen van het hartritme, ademhalingsproblemen of onderbrekingen tijdens de nachtrust.
Zwelling in de keel, een beklemd gevoel in de keel, stoornissen bij het slikken en veranderingen in vocalisatie worden als zorgwekkend beschouwd. Ze moeten door een arts worden onderzocht, zodat de symptomen kunnen worden verlicht. Als de betrokkene meer infecties lijdt, als het concentratievermogen en oplettendheid afneemt of als er herhaaldelijk navel- of liesbreuken optreden, moet een arts op de hoogte worden gebracht van de waarnemingen.
Plotselinge puistjes, gele verkleuring van de huid en innerlijke rusteloosheid moeten worden onderzocht en behandeld. Een arts is nodig zodra er pijn in het lichaam, verminderde kwaliteit van leven en gedragsproblemen zijn. Als er een risico op kortademigheid bestaat, is een ambulancedienst vereist. Om deze acute aandoening te voorkomen, moet zo snel mogelijk een arts worden geraadpleegd.
Therapie en behandeling
Een causale therapie is vandaag nog niet mogelijk. Er zijn echter enkele benaderingen voor toekomstige gentherapie voor deze ziekten in onderzoeksprojecten. Helaas zijn er momenteel geen tastbare resultaten op dit gebied. Er zal echter een klinische proef naar gentherapie voor de ziekte van Hurler beginnen in Barcelona. Bij sommige vormen van mucopolysaccharidose zijn beenmergtransfers in individuele gevallen effectief gebleken. Dit treft bijvoorbeeld de ziekte van Hunter, de ziekte van Hurler of de ziekte van Sanfilippo.
Door deze beenmergoverdracht worden de zieke stamcellen ingeruild voor gezonde stamcellen van een donor. Hierdoor kan het organisme het ontbrekende enzym voldoende herstellen. Enzymvervangende therapie loont in veel gevallen ook. Deze vervangingstherapie moet echter levenslang worden uitgevoerd. Er zijn echter ook gevallen waarin veelbelovende therapieën niet meer mogelijk zijn. Het gaat hier echter om symptomatische behandelingen.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen pijnOutlook & prognose
De verdere ontwikkeling bij patiënten met mucopolysaccharidose moet individueel worden beoordeeld. Deze term is een verzamelnaam voor verschillende stapelingsziekten. Deze zijn bij elke patiënt in verschillende mate aanwezig en hun intensiteit wordt individueel uitgesproken. Als er geen medische zorg wordt ingesteld, worden de inwendige organen van alle getroffenen in de loop van hun leven geleidelijk vernietigd. Dit resulteert in een verkorting van de gemiddelde verwachte levensduur.
Met een vroege diagnose kan een persoonlijk geoptimaliseerde therapie worden uitgewerkt. Dit hangt samen met de gezondheidseisen en bestaande klachten van de patiënt. Een langdurige behandeling is fundamenteel noodzakelijk om een stabiele verbetering van de gezondheid te bereiken. Er kunnen chirurgische ingrepen optreden, die elk met verschillende risico's en bijwerkingen zijn geassocieerd. Als de operatie zonder verdere complicaties verloopt, worden de symptomen meestal achteraf verlicht.
Toch kunnen er in de loop van het leven ongewenste ontwikkelingen en tegenslagen optreden. In individuele gevallen kan alleen een beenmergtransplantatie de algemene kwaliteit van leven verbeteren. Door de algemene omstandigheden ervaart de patiënt een sterke emotionele en mentale belasting. Door de symptomen is een normaal leven van alledag vaak niet mogelijk. Er kunnen psychische complicaties optreden die tot een verdere verslechtering van de situatie leiden.
preventie
Omdat mucopolysaccharidosen erfelijke ziekten zijn, is preventie niet mogelijk. In het geval van een bestaande ziekte kan het succes van de behandeling worden verzekerd door tijdige therapie. Bovendien is een constante controle van de long- en hartfunctie noodzakelijk. Als er zich al gevallen van mucopolysaccharidose in de familie hebben voorgedaan, kan het risico van de ziekte worden beoordeeld door middel van genetisch advies als de familie kinderen wil hebben.
Nazorg
In de meeste gevallen van mucopolysaccharidose heeft de patiënt maar weinig mogelijkheden voor nazorg, zodat de getroffene vooral in een vroeg stadium een arts moet raadplegen. Alleen met vroege opsporing en behandeling van deze ziekte kunnen verdere complicaties worden voorkomen, zodat een arts moet worden gecontacteerd zodra de eerste tekenen en symptomen optreden.
In de meeste gevallen zijn de getroffenen afhankelijk van chirurgische ingrepen, die de symptomen kunnen verlichten en beperken. Omdat mucopolysaccharidose echter een genetische ziekte is, kan deze meestal niet volledig worden genezen.
De betrokkene dient daarom eerst een arts te raadplegen als hij kinderen wil krijgen om herhaling van deze ziekte bij de kinderen te voorkomen. Vaak is het ook erg belangrijk om tijdens de behandeling gezinsondersteuning te hebben. Dit kan ook depressie en andere psychische stoornissen voorkomen. Mucopolysaccharidose kan leiden tot een verminderde levensverwachting van de getroffen persoon, waarbij het verdere verloop sterk afhangt van het tijdstip van diagnose.
U kunt dat zelf doen
De mogelijkheden voor zelfhulp bij mucopolysaccharidose zijn beperkt tot het verlichten van symptomen en daarmee het verbeteren van de kwaliteit van leven. Zelfhulpgroepen zijn zeer nuttig gebleken, aangezien de uitwisseling met andere ouders waardevolle tips oplevert en vaak angsten en zorgen kan verlichten en een positievere kijk op de toekomst kan geven.
De begeleiding bij fysiotherapie, ergotherapie, logopedie en andere vormen van therapie, die vaak in de thuissituatie kunnen worden verdiept, zijn nu een integraal onderdeel van het leven.
Om het leven voor uzelf en het getroffen kind zo gemakkelijk mogelijk te maken, is het raadzaam om de woonomgeving zo vroeg mogelijk toegankelijk te maken voor gehandicapten. Naarmate de leeftijd en het gewicht van het kind toenemen, blijken in hoogte verstelbare verpleegbedden een grote fysieke opluchting voor de verzorger. Epilepsiewaarschuwingsapparatuur en andere technische hulpmiddelen zorgen voor de best mogelijke veiligheid, zelfs 's nachts en ontlasten de ouders' s nachts, zodat ze meer ontspannen kunnen slapen.
Het bijhouden van een symptoomdagboek kan de arts helpen nieuwe symptomen te herkennen en mogelijk de behandeling van bestaande symptomen te corrigeren, aangezien medicamenteuze therapie vaak niet het gewenste effect laat zien, maar het tegenovergestelde effect.
Omdat de ziekte erg veeleisend is voor familieleden, moeten ze voor zichzelf weinig ruimte creëren om hun batterijen op te laden. Dit kan kuren, preventieve zorg of later een vakantie in het hospice zijn.





.jpg)
.jpg)

.jpg)