De Smith-Lemli-Opitz-syndroom is een cogenitaal misvormingssyndroom. De oorzaak is een van de in totaal 70 genmutaties op chromosoom 11q13.4. De ziekte wordt op een autosomaal recessieve manier overgeërfd en is een uiterst zeldzame ziekte met meerdere misvormingen van organen en een verminderde biosynthese van cholesterol.
Wat is het Smith-Lemli-Opitz-syndroom?

© jijomathai - stock.adobe.com
De Smith-Lemli-Opitz-syndroom valt in de groep van autosomaal recessieve erfelijke misvormingssyndromen. Een genmutatie veroorzaakt een stofwisselingsstoornis in de biosynthese van cholesterol bij de ziekte. Het syndroom is de meest voorkomende cogenitale aandoening van de biosynthese van cholesterol. De prevalentie van de ziekte in Europa ligt tussen ongeveer 1: 60.000 en 1: 10.000.
$config[ads_text1] not found
De ziekte kan dus worden geclassificeerd als een zeldzame ziekte, hoewel het een van de meest voorkomende cogenitale ziekten is bij de biosynthese van cholesterol. Het syndroom is nog zeldzamer op de continenten Azië en Afrika. De ziekte werd voor het eerst beschreven in 1964. De genetici D. W. Smith, L. Lemli en J. Marius Opitz documenteerden het symptoomcomplex vanuit wetenschappelijk oogpunt. Sindsdien zijn iets meer dan 300 gevallen gemeld.
Jongens worden vaker getroffen dan meisjes. De symptomen zijn waarschijnlijk meer uitgesproken bij meisjes en daardoor moeilijker te diagnosticeren. De ziekte is aangeboren, maar ontwikkelt zich vanaf de geboorte geleidelijk en wordt daarom gekenmerkt door verschillende vormen. Het syndroom is onderverdeeld in type I en II, afhankelijk van de symptomen.
oorzaken
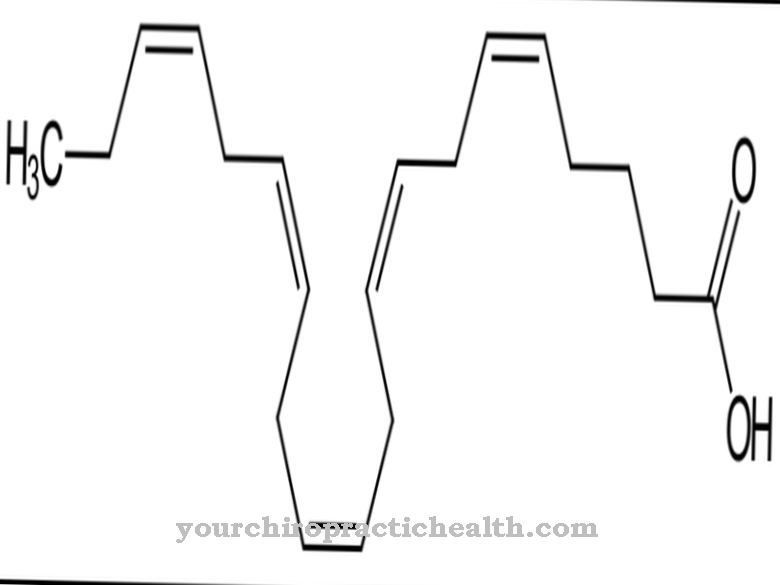
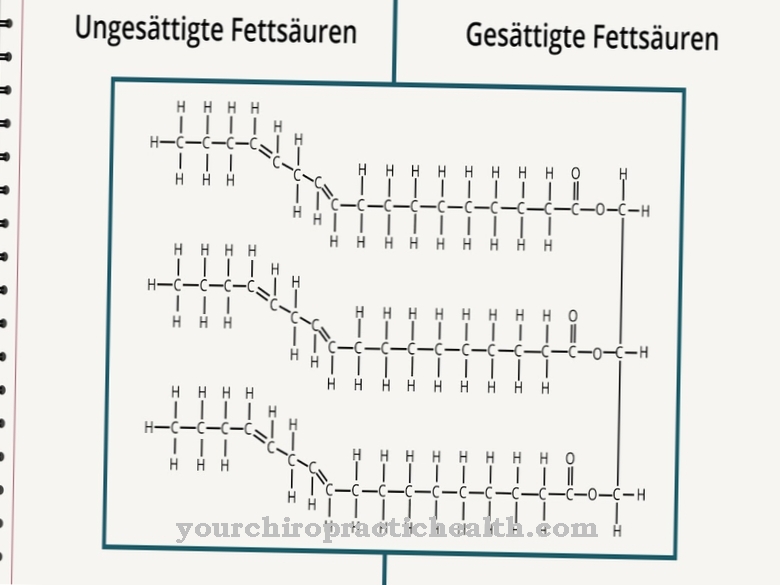
Smith-Lemli-Opitz-syndroom wordt veroorzaakt door een genmutatie die in 1998 werd gelokaliseerd. Chromosoom 11q13.4 wordt nu beschouwd als de locatie van de mutatie, met tot op heden meer dan 70 verschillende mutaties op deze locatie bekend. Het type causale mutatie bepaalt de ernst en het type symptomen in elk individueel geval. Het gen in kwestie is het 7-sterol-reductasegen. S. Tint ontdekte samen met zijn collega's dat het syndroom de eigen aanmaak van cholesterol door het lichaam verhindert.
$config[ads_text2] not foundDeze productie omvat de omzetting van de precursor 7-dehydrocholesterol in het lichaamseigen cholesterol, dat niet kan functioneren vanwege een enzymdefect als gevolg van de mutatie. Er is dus een teveel aan 7-dehydrocholesterol in het lichaam van de getroffenen. Tegelijkertijd is er een algemeen tekort aan cholesterol. Vanwege de autosomaal recessieve overerving van het syndroom, moeten beide ouders het defecte gen dragen en kunnen ze het alleen op deze manier aan een kind doorgeven. De kans is 25 procent dat de volgende kinderen van ouders met een ziek kind ook door het misvormingssyndroom worden getroffen.
Symptomen, kwalen en tekenen
Kinderen met het Smith-Lemli-Opitz-syndroom worden geboren met typische craniofaciale misvormingen, vooral microcefalie, een prominent voorhoofd en een kleine neus met een brede neuswortel. Naast een anteverted neusgaten is er een microgenie. Een gespleten gehemelte en vertroebeling van de lens worden ook vaak waargenomen, vooral cataracten en cataracten.
Blepharoptosis is ook aanwezig. Mentaal en cerebraal ontwikkelen zich in de loop van de tijd ongewenste ontwikkelingen die resulteren in een verstandelijke handicap. Holoprosencefalie en prikkelbaarheid kunnen ook het klinische beeld bepalen. Naast zelfbeschadigend gedrag kan het syndroom autistisch gedrag uitlokken. Bovendien zijn er meerdere misvormingen van organen, vooral die van het hart en de urogenitale tractus.
$config[ads_text3] not foundHypospadie en cryptorchisme zijn de meest voorkomende urogenitale misvormingen. Naast overtollige vingers of tenen kan teensyndactylie aanwezig zijn. Spierhypotonie, slikstoornissen en gastro-oesofageale reflux zijn ook relevant in de context van het symptoomcomplex. Intestinale dysmotiliteit en pylorusstenose komen ook vaak voor. Bij type II van het syndroom is er pseudo-hermafroditisme, waarbij de uitwendige geslachtsorganen vrouwelijk zijn, hoewel het mannelijke karotype overheerst.
Diagnose en ziekteverloop
Als prenatale diagnose kunnen echografie-onderzoeken de typische fysieke kenmerken van het Smith-Lemli-Opitz-syndroom al vóór de geboorte registreren. Naast een groeistoornis kan een hartafwijking of het ontbreken van een nier worden opgemerkt. Bij de vruchtwatertest kan de mutatieanalyse al een diagnosebevestigend resultaat opleveren.
Na de geboorte hebben de kinderen een karakteristieke gezichtsvorm en speciale posities van de ledematen, zodat de vermoedelijke diagnose kan worden gesteld door visuele diagnose als de prenatale diagnose is mislukt. De genetische diagnose stelt het vermoeden veilig. In termen van differentiële diagnose moet het Smith-Lemli-Opitz-syndroom worden onderscheiden van het foetaal alcoholsyndroom, het Pallister Hall-syndroom, het Kaufmann-McKusick-syndroom en het Cornelia de Lange-syndroom.
Het Pätau-syndroom, het ATR-X-syndroom en het C-syndroom, het Zellweger-syndroom en het hydrolethalussyndroom moeten ook in de differentiële diagnose worden meegenomen. Hetzelfde geldt voor het Oro-Faciales-Digitales Syndroom, het Holoprosencefalie-Polydactylie Syndroom en het Meckel-Syndroom. De levensverwachting van kinderen hangt af van de cholesterolconcentratie en de behandelbaarheid van de orgaanmisvormingen. Lage cholesterolwaarden en de meest ernstige misvormingen leiden tot een spoedig fatale afloop. Kinderen met een hoog cholesterolgehalte en gemakkelijk te behandelen misvormingen hebben geen ernstig verminderde levensverwachting.
$config[ads_text4] not found
Complicaties
Vanwege het Smith-Lemli-Opitz-syndroom lijden de getroffenen aan verschillende misvormingen en misvormingen. Deze hebben een zeer negatief effect op de kwaliteit van leven van de patiënt. Met name de inwendige organen worden door de misvormingen aangetast, waardoor de dood vaak direct na de geboorte kan optreden. Bovendien hebben de meeste patiënten last van een gespleten gehemelte en ook van oogproblemen.
Bovendien leidt dit syndroom vaak tot verstandelijke beperkingen en dus tot verstandelijke achterstand. De meeste patiënten zijn daarom in hun leven afhankelijk van de hulp van andere mensen en kunnen veel alledaagse taken niet meer alleen aan. Het hart wordt ook beïnvloed door de misvormingen, die kunnen leiden tot plotselinge hartdood. Verder tast het Smith-Lemli-Opitz-syndroom ook de geslachtsorganen aan, zodat ook hierin misvormingen kunnen optreden.
Behandeling van het Smith-Lemli-Opitz-syndroom kan gewoonlijk alleen symptomatisch zijn. Er zijn geen complicaties en sommige symptomen kunnen beperkt zijn. Een volledig positief verloop van de ziekte kan echter niet worden bereikt. Of de levensverwachting beperkt zal zijn, is niet universeel te voorspellen.
Behandeling en therapie
Voor patiënten met het Smith-Lemli-Opitz-syndroom is levenslange sociale en medische zorg vaak onvermijdelijk. In de regel loopt hun ontwikkeling ernstig vertraagd op cognitief en motorisch gebied. Dit resulteert in bijna alle gevallen in een levenslange handicap die een zelfstandige levensstijl niet toelaat. Daarom wordt vooral ondersteunende behandeling geboden.
Als onderdeel van deze maatregelen krijgen ouders psychotherapeutische ondersteuning en leren ze idealiter om te gaan met de ziekte van hun kind. Het Smith-Lemli-Opitz-syndroom is ongeneeslijk en kan daarom niet als oorzaak worden behandeld. Aangezien voor het syndroom een stoornis van het cholesterolmetabolisme is gedocumenteerd, is symptomatische behandeling ter compensatie van het cholesteroltekort mogelijk. Deze behandeling wordt gedaan door cholesterol te geven.
De veelal talrijke misvormingen van de organen moeten voor zover mogelijk operatief worden gecorrigeerd. Een uitzondering hierop is de veelvuldig gedocumenteerde meervoudige koppeling van de vingers en tenen, waarvoor geen chirurgische ingreep nodig is. Bijkomende symptomen zoals visuele problemen kunnen nu ook goed worden behandeld en verlicht.
De meerderheid van de getroffenen lijdt aan voedingsproblemen zoals zuigen en slikken, of gastro-oesofageale reflux en verminderde gastro-intestinale peristaltiek. Daarom moet vaak een maagsonde worden gebruikt om een veilige voedselopname te garanderen. Gedragsproblemen kunnen mogelijk worden behandeld met gedragstherapie.
preventie
Na een positieve prenatale diagnose voor het Smith-Lemli-Opitz-syndroom krijgen ouders de mogelijkheid om de zwangerschap te beëindigen. Het Smith-Lemli-Opitz-syndroom kan alleen op een andere manier worden voorkomen als paren een moleculair genetische diagnose laten stellen in hun gezinsplanning en, nadat bewijs is geleverd over de mutatie, hun eigen kinderen afwijzen.
Nazorg
De vervolgmaatregelen voor het Smith-Lemli-Opitz-syndroom (SLOS) zijn gebaseerd op de ernst van de symptomen die optreden tijdens het ziekteverloop. In de meeste gevallen van ziekte hebben de kinderen voedingsproblemen. Ze doen het slecht. De focus van de nazorg ligt daarom allereerst op de adequate voeding van de getroffen kinderen door middel van nasogastrische calorierijke vloeibare voeding en het toedienen van voldoende cholesterol.
Het verdere verloop van de ziekte toont ook bij veel van de getroffen kinderen een onderontwikkeling van de hersenen. Deze onderontwikkeling leidt gewoonlijk tot lichamelijke of geestelijke handicaps met verschillende gradaties van ernst. Zo leren niet alle getroffen kinderen lopen. Ter compensatie van de beperkte mobiliteit worden in dit geval hulpmiddelen voor alledaagse bewegingen (bijv. Rolstoel-, loop- en stahulpmiddelen) verstrekt als vervolgmaatregel.
Bij mentale symptomen zoals autoagressie en hyperactiviteit wordt de therapeutisch voorgeschreven medicamenteuze behandeling als vervolgmaatregel voortgezet. Bovendien zal ongeveer 50 procent van alle getroffen kinderen een matig tot ernstig hartafwijking ontwikkelen naarmate de ziekte voortschrijdt. Na de operatie van het hartafwijking worden regelmatig elektrocardiografische (ECG) en sonografische onderzoeken gepland.
Voor ouders van kinderen met SLOS worden psychologische consulten en therapieën aanbevolen. Zelfstandig leven op volwassen leeftijd is nogal onwaarschijnlijk. Uitgebreide zorgmaatregelen zijn te verwachten in de nazorg van de SLOS op volwassen leeftijd. Bovendien kunnen orgaanmisvormingen de levensverwachting beperken.
U kunt dat zelf doen
De ziekte gaat gepaard met tal van klachten die de kwaliteit van leven ernstig aantasten. Als bij een familielid de genetische ziekte is vastgesteld, moet een arts worden geraadpleegd voordat het kind wordt verwekt. Mogelijke risico's moeten worden afgewogen, zodat voor alle betrokkenen prudente beslissingen kunnen worden genomen. Bovendien moet u deelnemen aan alle controles die tijdens de zwangerschap worden aangeboden.
Zodra de gezondheidsproblemen van het kind worden opgemerkt, kunnen ouders en familieleden passende voorzorgsmaatregelen nemen en zich beter voorbereiden op toekomstige ontwikkelingen. In een groot aantal gevallen is de zorg voor iemand met het Smith-Lemli-Opitz-syndroom een enorme uitdaging voor de nabestaanden. Daarom moeten ze hun fysieke en emotionele grenzen kennen en naleven. Het is raadzaam om het gebruik van medische zorg voor de patiënt en, parallel, psychotherapeutische ondersteuning voor de naasten te overwegen. Als gevolg hiervan kunnen vaak verbeteringen worden bereikt bij het omgaan met de ziekte.
Opgemerkt moet worden dat stabiliteit van de sociale omgeving belangrijk is voor de patiënt. Bovendien moet u altijd kalm blijven in het dagelijks leven als zich tegenslagen en uitdagingen voordoen. Aangezien de persoon in kwestie niet in staat is zijn leven zelfstandig vorm te geven, moet er een speciale empathie zijn in de omgang met hem.

.jpg)
.jpg)
.jpg)




.jpg)